30 Jan 2015
Today we’re excited to announce the release of Guacamole, a variant calling platform that can run an order of magnitude faster than existing solutions and allows for faster iteration on variant calling algorithms.
Background
Variant calling, the process of identifying differences between two or more genomes, is an important stage in many genome analysis pipelines.
Human genomes are commonly sequenced as billions of overlapping short reads that are aligned in ways that maximize agreement about the underlying base pairs, but the data is typically noisy enough that many positions show evidence of being two or more values:
(A, C, or T?)
In addition, at hundreds of gigabytes of data per genome, a typical variant caller only attempts to process a few genomes and can take hours to run. This has slowed the development of better variant callers; who wants to tweak parameters and experiment on algorithms with a turn-around time of hours?
As a result, a few callers have become de facto standards, but they weren’t designed to scale to thousands (let alone millions) of genomes and, worse, there is mounting evidence that they diverge in their output (for fixed inputs), sometimes wildly!
All downstream treatment decisions hinge on the variants discovered in patients’ genomes, so it is crucial that we gain better understanding of how to quickly and accurately identify variants.
Guacamole
With this in mind, we developed Guacamole, which makes it easy to implement variant calling algorithms and run them on modern, distributed infrastructure. Along with Cycledash (our variant visualizer) and Ketrew (our workflow manager), we’re able to iterate on variant callers, run them, and view their output easily and in a matter of minutes.
- Three reference variant callers, all expressed in ~100-200 lines of code and that run on full genomes in as few as 20 minutes:
- “Germline standard” caller: emits any variants whose likelihood (based on the quality of the reads displaying them) clears a certain threshold.
- “Germline threshold” caller: emits variants that appear in above a certain percentage of the reads.
- “Somatic standard” caller: compares a “tumor” sample to a “normal” sample (both having been aligned to the same reference) and emits likely variants that are specific to the former (e.g. mutations found in a cancer tumor).
- Primitives for performing genomic analysis:
partitionLociByApproximateDepth:
- Assigns genomic regions to processing tasks, balancing the number of reads that align to each region.
- High coverage regions are divided into many small partitions; low coverage regions are merged.
pileupFlatmap:
- Run a user-specified function on each locus of the genome, passing it a pileup giving the relevant bases from all reads mapping to that locus.
windowFlatMap:
- Run a user-specified function on sliding windows of a specified size in the genome.
- All reads mapping within a window centered around each locus are passed to the user function.
- Like
pileupFlatmap, but for algorithms that require additional context around each locus.
What Are We Doing With It?
- We used Guacamole to enter variant calls into the recent DREAM Somatic Mutation Calling Challenge.
- We’ve also begun to tailor it to various clinical applications, including the development of precision cancer immunotherapies with collaborators from Mt. Sinai and Memorial Sloan Kettering.
Limitations / Roadmap
The callers included in 0.0.0 are experimental; we don’t recommend using them in production yet. Below are some things we are working on adding or improving.
Structural Variants
While the SNV-calling functionality in Guacamole is fairly robust, the indel- and SV-calling functionality is still fairly naïve.
- Better structural-variant calling is being worked on.
- We also plan to allow Guacamole to ingest reads from multiple sequencing platforms, e.g. longer PacBio reads, and make calls based on “hybrid” short- and long-read data.
Scalability
Spark, which we use as our distributed runtime, is difficult to debug and frequently fails on inputs larger than a few hundred GB (compressed; this can end up being tens of TB in deserialized-Scala-object form).
- We have built a prototype variant caller using the Crunch/Scrunch interfaces to Hadoop’s implementation of MapReduce to establish that these algorithms do complete when run on more robust runtimes.
- We are actively working on Spark’s monitoring capabilities.
- Building a pipeline capable of rapidly analyzing genomic data from hundreds or thousands of patients is a high priority.
Clinical Applications
Guacamole will be in used in various downstream applications in the coming months:
- An upcoming cancer vaccine trial will use somatic variant calls from Guacamole (in conjunction with other callers) to identify candidate biomarkers in patients’ cancer cells.
- We are studying concordance between existing variant callers, Guacamole included, in an effort to better characterize the failure modes of current approaches.
You can find more info about Guacamole, as well as see what we’re working on and file issues, on GitHub.
28 Jan 2015
When iterating on a variant caller,
it’s helpful to be able to rapidly identify the most interesting variants called
and to visually inspect the evidence used by the caller to make each call.
CycleDash is our variant call analysis
application, used to inspect, query, compare, and export variant calls imported
from VCF files.
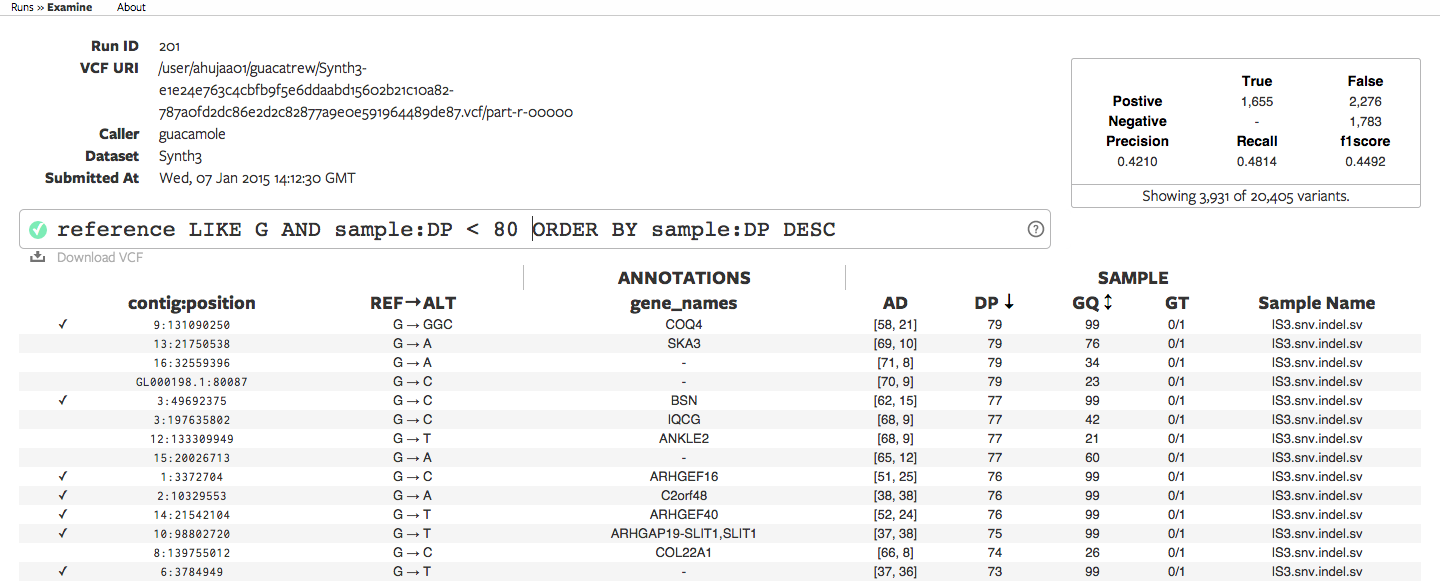
CycleDash displays variant calls in a tabular format. It provides summary statistics,
annotations (e.g. the gene within which a variant occurs), a discussion/commenting
system, and a powerful query omnibar which allows the user to quickly filter and
sort the variants using an SQL-inspired syntax. It allows users to export the
filtered variants as a VCF file for further downstream analysis.
Users can manually upload a VCF file to CycleDash through the web interface or they
can point CycleDash to a VCF file stored on HDFS.
When a VCF is submitted through either method, a request to process the VCF is placed on a work queue.
Once processing is complete, the processed data is inserted into a PostgreSQL database
in a denormalized format.
The variants are then available for inspection through the web interface.
What’s in 0.0.0?
- A RESTful JSON API for submitting and querying VCFs.
- A dashboard listing all submitted VCFs + metadata.
- An “Examine” page for filtering and exploring the variants in a VCF, including
- A SQL-like query language for filtering and sorting variants.
- Download functionality for saving queried variants.
- An embedded pileup viewer for viewing variants in context.
- Summary statistics compared to a validation VCF, e.g. precision, recall,
and F1 score.
- Workers for processing and annotating VCFs.
- A comment system for discussing variants.
What Are We Doing With It?
We’ve primarily used CycleDash to debug and improve Guacamole, our
somatic variant caller. Ketrew posts the results of a run of
Guacamole (or any other variant caller) to CycleDash,
which in turn processes and imports the variant calls into PostgreSQL, where they
are stored for later inspection.
In the future, we would like to use CycleDash to explore the functional consequences of variants.
Exploring, discussing, and analyzing the results of a variant calling
pipeline is often done in a series of Excel spreadsheets, written notes, and
static images. When mature, CycleDash may be an improvement on existing workflows.
Current Limitations
CycleDash currently has trouble querying and serving call sets with over a million
variants. We hope to improve performance for large call sets by optimizing our SQL queries.
If we are not able to obtain the desired performance with PostgreSQL, we are open to exploring
other databases. It’s a priority that CycleDash be responsive for even
the most complex queries over the largest call sets.
Queryability
Our query engine cannot yet execute queries like “find all true positives”,
“show all indels”, and doesn’t yet support queries over annotations or
comments. We are also planning to support aggregate queries, user-defined
functions, and saved queries.
Robust Error Handling
CycleDash fails in odd ways when it gets malformed and unexpected input. This is
very confusing for users, and often involves diving into logs to figure out how
something went wrong. Better error reporting is part of our ongoing goal to
offer a seamless and pleasant UX to developers and non-technical users alike.
Roadmap
Some other highlights of our roadmap include the following:
- Comparison and concordance between different call sets.
- Introducing histograms and other visualizations and statistics.
- More annotation options and better pluggability for adding new annotations.
- A new, more performant, and purpose-built pileup viewer, pileup.js.
- Links between variants in CycleDash and the UCSC Genome Browser.
You can track (and add to) the project’s issues & features on
GitHub Issues.
23 Jan 2015
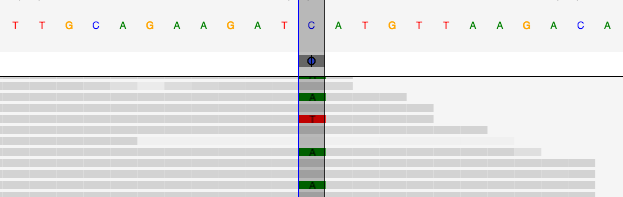
One of the key features of CycleDash is the embedded pileup view,
currently provided by BioDalliance. This lets you inspect the reads which
contributed to a variant call:
 (This pileup shows a single nucleotide variation between the top sequence (C)
and the bottom sequence (T), relative to a reference genome (C).)
(This pileup shows a single nucleotide variation between the top sequence (C)
and the bottom sequence (T), relative to a reference genome (C).)
We noticed that the two pileup tracks were quite slow to load, often taking in
excess of 30 seconds to appear. This latency discouraged use of the feature, so
we set out to improve it.
Pileup data is typically stored in a BAM file, which encodes all the
short reads in the order in which they appear in the genome. Because genomes
are large and sequenced at high read depth, BAM files can be quite large.
For example, the synth3 BAM files from the DREAM Somatic Mutation
Calling Challenge weigh in at 177.4GB each.
Transferring such a large file to a user’s web browser is impractical. Instead,
BioDalliance (and other genome browsers such as IGV) load a BAM
Index (BAI), which they then use to load only small portions of the
larger BAM file.
As BAM files grow, so do their corresponding BAI index files. For the synth3
data set, the two BAI files are each 8.6MB (4.9MB compressed). This is still quite a bit of
information to transfer to a web browser, and it accounted for the slow load
time that we were seeing:

(The two BAI files block the loading of other resources for seven seconds.)
We tried some standard tricks like applying GZip compression to the BAI files,
but the size of these files remained a fundamental barrier to interactive use.
In principle, only a tiny portion of the BAI file is needed at any given time.
For example, if the user wants to look at Chromosome 20, then there’s no need
to load index data for Chromosome 3.
After some discussion on BioStars and the samtools-devel
mailing list, we decided to index the BAM index! This results in a
small JSON file which records the beginning and ending of each contig in the
BAI file (“contig” is a general term for a contiguous stretch of DNA, e.g. a
chromosome):
{
"chunks": [
[8, 716520], // byte offsets for Chromosome 1
[716520, 1463832], // byte offsets for Chromosome 2
[1463832, 2070072], // etc.
...
]
}
We call this an “index chunk file” because it records the locations of
different chunks of the BAM Index. For example, the above file records that
Chromosome 3 begins at byte 1,463,832 in the BAI file and extends through byte
2,070,072. To display pileups from Chromosome 3, a genome viewer need only load
these ~600K of the BAI file, rather than the full 8.6M. This results in a
considerable speedup:

The overall load time has been reduced from 10s to something like 3s.
You might wonder, what happens if this index of the index gets too
large? For a human genome, it’s quite small (1.7KB for synth3) and can be
inlined into the page or requested separately. And its size depends only on the
number of contigs, not the number of reads, so it won’t grow with sequencing
depth (like a BAI file does).
To incorporate BAI indices into your own software, you’ll want to look at the
bai-indexer Python project and the change to BioDalliance
which added support. We typically store the index chunks in a .bai.json file
alongside the standard .bam and .bai files, so they never get separated.
While we’d hoped not to add new files and formats to an already large
family, we found that it was impossible to incorporate higher level
indexing data into the BAI file itself. By using a JSON format, we stay
flexible to future improvements and extensions to this scheme.
By creating a higher level index, we were able to significantly improve the
load time of genomic visualizations. We’d love to see wider support for this
convention, so check out the bai-indexer project!
21 Jan 2015
We just released Ketrew 0.0.0 in
opam’s main repository.
Ketrew means Keep Track of
Experimental Workflows: it is a workflow engine based on an Embedded Domain
Specific Language.
- The EDSL is a simple OCaml library providing combinators to:
- define complex workflows (interdependent steps/programs using a lot of
data, with many parameter variations, running on different hosts with
various schedulers)
- submit them to the engine.
- The engine orchestrates those workflows while keeping track of everything
that succeeds, fails, or gets lost.
Ketrew can be run as a standalone application (i.e. run the engine from the
command line), or using a client-server architecture (i.e. run a proper server,
and connect through HTTPS).
What’s in 0.0.0?
- An EDSL that can be used to build complex workflows: we wanted to keep the
EDSL usable by OCaml beginners (read: it is not a bunch of
GADTs carrying
1st class modules).
- Client-server (HTTPS + JSON) and standalone modes.
- An implementation of the engine which is naive/slow but testable &
understandable.
- A command-line client that can explore the state of workflows and pilot the
engine interactively.
- A persistence layer based on an “explorable” (but slow)
git database.
- Access through SSH to LSF and PBS-based computing clusters, and
also to Unix machines that do not have a batch scheduler.
- A plugin infrastructure to add backends (to be linked with the library or
using
Dynlink and/or Findlib (cf.
documentation).
What We Are Doing With It
Ketrew has been used for very diverse things like running backups or building
documentation websites (see
smondet/build-docs-workflow).
But we actually develop and use Ketrew to run bioinformatics pipelines on
very large amounts of data:
- Biomedical computational pipelines involve various long-running computational
steps; for each of them, we want to run many parameter variations.
- At the same time, bioinformatics tools are infamous for being quite poorly
engineered; they tend to fail in mysterious ways, for seemingly valid inputs.
- The computing infrastructure is also very diverse and gives little control to
the end user.
Ketrew is designed with those adverse conditions in mind.
By the way, we just started open-sourcing our Ketrew pipelines for genomics
experiments, see hammerlab/biokepi: it
is a target library (i.e. a set of functions to create Ketrew.Target.t
values which wrap the installation and running of bioinformatics tools), but it
also contains a cool GADT-based pipeline description module; we’re on the road
to concise and well-typed bioinformatics pipelines!
Limitations & Future Work
We are switching to faster database backends: see the library
smondet/trakeva (see PR
#98).
We had to slow down Ketrew quite artificially because submitting too many
targets (≥ 100-ish), that all check-up on files/conditions on the same SSH host,
would cause connection errors, we will work on less naive approaches for
accessing remote hosts (see issue
#69).
We are also working on the engine itself, see issue
#73.
(G)UI
We routinely successfully run workflows that contain more than 1000 steps each,
but we need better user-interfaces to go much further (both web- and
command-line-based even emacs/vim friendly).
Deployment
Because of ocp-build, Ketrew 0.0.0 requires OCaml 4.01.0; we are switching
to an ocamlbuild/oasis -based build while waiting for
Assemblage to be ready.
We hope that you will find Ketrew useful for any of the various kinds of
workflows you may want to track! Feel free to file issues or reach out at
hammerlab/ketrew if you have any
questions.
If you are in New York City by the end of January, we will be at the
Compose conference to present Ketrew.
05 Dec 2014
Genomic data sets can be quite large, with BAM (binary alignment) files easily weighing in at hundreds of gigabytes or terabytes for a whole genome at high read depth. Distributed file systems like HDFS are a natural fit for storing these beasts.
This presents interactive genomic exploration tools like IGV with a challenge: how can you interact with a terabyte data file that may be on a remote server? Downloading the entire file could take days, so this isn’t an option.
IGV and other tools create interactive experiences using a few tricks:
- They require a BAI (BAM Index) file, which lets them quickly determine the locations in the BAM file which contain reads in a particular range of interest.
- They set the rarely-used
Range: bytes HTTP header to request small portions of the BAM file from the remote web server.
In practice, this means that IGV can display reads from any location in a 100 GB BAM file while only transferring ~100KB of data over the network.
Popular web servers like Apache and nginx support the Range: bytes header, but WebHDFS, the standard HTTP server for content on HDFS, does not. It does support requesting ranges of bytes in a file, however, but it does so using its own URL parameters. IGV does not know about these parameters, so it can’t speak directly to WebHDFS.
We can solve this problem by building an adapter. And that’s exactly what igv-httpfs is.
igv-httpfs exports a WSGI server which understands Range: bytes HTTP headers and converts them to appropriate HTTP requests to WebHDFS. This allows IGV to load files from HDFS.
igv-httpfs also sets appropriate CORS headers so that web-based genome visualizations such as BioDalliance which make cross-origin XHRs can enjoy the same benefits.
As with any WSGI app, we recommend running igv-httpfs behind a WSGI HTTP server (we use gunicorn) and an HTTP server (we use nginx). This improves performance and lets you enable gzip compression, an essential optimization for serving genomic data over the network.
Update (2015-12-14): Another approach to serving HDFS content over HTTP is to create an NFS mount for the HDFS file system, say at /hdfs. With this in place, you can serve HDFS files using a standard HTTP server like nginx or Apache. Some care is required to correctly support CORS, caching and bioinformatics MIME types, but support for range requests comes built-in. You can use our nginx configuration as a template.
